


La litiasis urinaria es una patología de origen multifactorial, en la que están implicados factores ambientales (climáticos), dietéticos, metabólicos y relacionados con el estilo de vida. Entre ellos también se encuentran los genéticos. Estos pueden ser poligénicos (mutación patogénica en diferentes genes) o monogénicos (mutación patogénica en un único gen).
La litiasis urinaria afecta a un amplio sector de la población, entre el 4 y el 15% aproximadamente y, es una de las patologías que causa mayor repercusión en la calidad de vida y en la actividad laboral o cotidiana durante su fase aguda (dolor cólico). Entre el 10 y el 15% de los cálculos urinarios requieren tratamiento activo y, entre el 20 y el 30% hospitalización. La tasa de recurrencia es de un 50-70% a 5 años si no han llevado o cumplimentado el tratamiento preventivo adecuado.
La litiasis en la edad pediátrica es menos frecuente con una incidencia de 0,13–1,52 casos por cada 1000 admisiones hospitalarias. La incidencia de litiasis infantil se ha multiplicado por cinco en las últimas décadas y, la modificación de los patrones alimenticios es una de las principales causas [1]. La litiasis infantil es especialmente relevante debido a la posibilidad de causar lesiones estructurales en el riñón [2].
El conocimiento de las causas que han originado el episodio litiásico es esencial para el correcto diagnóstico y tratamiento de la enfermedad.
La influencia de la carga genética parece jugar un importante papel en la enfermedad litiásica, de modo que generalmente entre el 45% y el 55% de los casos presentan antecedentes familiares (especialmente en aquellos pacientes en los que subyace una alteración metabólica). La hipercalciuria y la hipocitraturia son las alteraciones metabólicas más frecuentes en los pacientes con litiasis urinaria. La forma más frecuente de hipercalciuria es la idiopática. La hipercalciuria idiopática es la situación clínica en la que hay un incremento en la eliminación urinaria de calcio, en ausencia de hipercalcemia y de otras causas conocidas de hipercalciuria.
La hipercalciuria idiopática es una condición multifactorial y poligénica que en muchos casos se transmite de forma autosómica dominante y se relaciona con variantes en diversos genes (single nucleotide polymorphisms o SNPs) que pueden modular la calciuria o la cristalización de sales cálcicas. Así por ejemplo se han descrito SNPs en CASR (receptor sensor de calcio) o en VDR (receptor de vitamina D) [3] [4] [5]. Sin embargo, parece que otros genes intermediarios también podrían estar implicados [6].
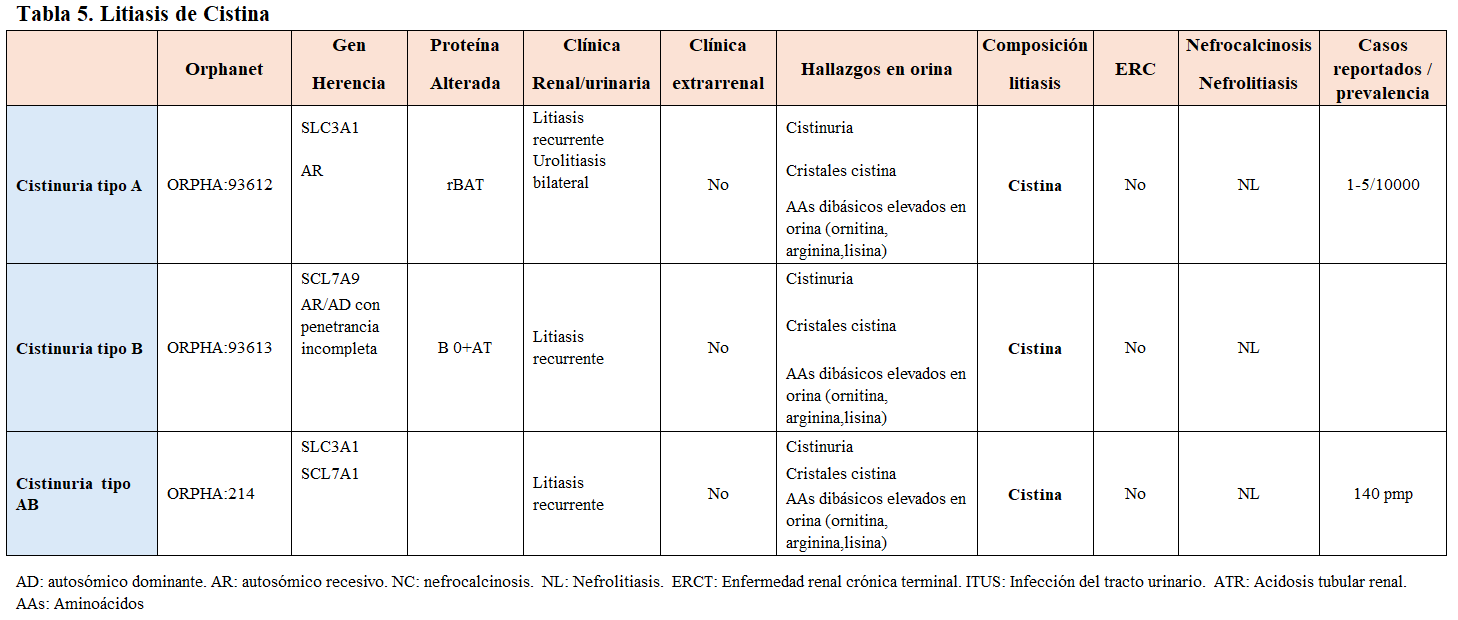
La frecuencia de cálculos urinarios causados por una alteración de causa monogénica es baja (entorno a un 2%), sin embargo, otras revisiones llevadas a cabo en centros de referencia de patología litiásica infantil y de adultos (stone centers), la sitúan en un 15-30% [7] [8] [9]. Como ejemplo de enfermedad litiásica monogénica, la cistinuria es la más frecuente en niños y, puede llegar a representar el 10% de las litiasis renales infantiles. Constituye la causa monogénica de enfermedad renal litiásica más frecuente en niños [10].
Al menos, se han identificado 40 genes relacionados con la litiasis urinaria o la nefrocalcinosis (depósito de calcio en el riñón). Muchas de estas patologías monogénicas que causan litiasis urinaria se deben a tubulopatías primarias o hereditarias, es decir anomalías de la función del túbulo renal. Dependiendo de la función tubular que se encuentre afectada, cada entidad tiene una edad de aparición, unas manifestaciones clínicas y analíticas y, una gravedad y pronóstico propios. La clínica general cuando debutan en la infancia suele ser la presencia de astenia, vómitos, retraso de crecimiento, sed y poliuria, acompañado de alteraciones electrolíticas y manifestaciones extrarrenales como alteraciones oculares o hipoacusia [11] [12].
Otras patologías que pueden causar litiasis urinaria y que se producen por alteraciones genéticas son los errores innatos del metabolismo como las hiperoxalurias primarias. Se deben a mutaciones en diferentes tipos de enzimas hepáticos que conllevan a la no degradación del oxalato con la consecuente eliminación excesiva por orina (hiperoxaluria). Estos niveles elevados de oxalato vencen los mecanismos de excreción renal y hace que se depositen en el riñón produciendo nefrolitiasis e incluso oxalosis sistémica (depósito de oxalatos en otros órganos, como esqueleto, corazón o hígado).
Dada la baja frecuencia de estas entidades y su variabilidad clínica, en ocasiones, el diagnóstico puede retrasarse años, lo que condiciona una peor calidad de vida y pronóstico para el paciente [13]. Es importante pensar en estas alteraciones para diagnosticarlas y poder instaurar un tratamiento precoz que prevenga o retrase el daño renal y la necesidad de tratamiento sustitutivo renal.
El objetivo de este artículo es hacer una revisión de la mayor parte de las patologías de origen genético que pueden condicionar la formación de litiasis urinaria y, definir el perfil clínico que precise un análisis genético [14].
Enfermedad litiásica. Clínica y diagnósticoLas manifestaciones clínicas y el diagnóstico de la enfermedad litiásica de origen genético no difieren de la enfermedad de origen no genético.
Manifestaciones clínicasLa manifestación clínica más frecuente de la litiasis urinaria es un dolor de inicio brusco, de tipo cólico, que va incrementándose paulatinamente y que se irradia de forma característica a fosa ilíaca, labios mayores o testículos ipsilaterales (cólico nefrítico). Puede asociarse a agitación psicomotriz, crisis hipertensiva arterial, polaquiuria, urgencia miccional o síntomas digestivos como nauseas o vómitos. En ocasiones, puede ser un hallazgo incidental en el contexto de un estudio con pruebas de imagen por otra patología. En los niños, la manifestación clínica suele ser diferente, con síntomas inespecíficos como dolor abdominal, náuseas o vómitos o incluso irritabilidad.
DiagnósticoEl diagnóstico de la enfermedad litiásica se realiza mediante pruebas de imagen, principalmente radiografía simple de aparato urinario, ecografía renal o una TC sin contraste. Algunas litiasis tienen particularidades, como las de 2,8 dihidroxiadenina, xantina y ácido úrico que son radiotransparentes, lo que puede dificultar su diagnóstico.
En el proceso diagnóstico, el análisis del cálculo urinario es el primer paso para la evaluación metabólica de un paciente formador de litiasis. Un adecuado análisis de la litiasis es fundamental para iniciar el proceso diagnóstico que nos pueda orientar hacia una alteración metabólica subyacente. La técnica ideal para el análisis del cálculo es la difracción de rayos X o la espectroscopia por infrarrojos. El análisis bioquímico del cálculo ha quedado relegado al no precisar de forma exacta la composición del cálculo. En centros especializados, la microscopía electrónica puede aportar información adicional sobre la historia de formación del cálculo, al poder visualizar y describir la forma de agregación de los cristales.
Tras el análisis del cálculo se debe realizar un estudio metabólico en sangre y orina en el que se pueda analizar la pérdida de equilibrio entre sustancias promotoras de la cristalización (calcio, fosfato, ácido úrico, oxalato y cistina) y sustancias inhibidoras (fundamentalmente citrato y magnesio) [15]. La evaluación metabólica no está indicada en pacientes de edad media, que presentan por primera vez una litiasis cálcica, por su bajo riesgo de recurrencia. Pero sí está indicado en niños, litiasis recidivantes, litiasis unilateral múltiple, litiasis bilateral, monorrenos, litiasis úricas, litiasis de cistina, y en pacientes con enfermedad endocrina, metabólica, ósea y gastrointestinal. El estudio metabólico se basa en determinaciones en orina de 24 horas de los parámetros implicados en la litogénesis [16].
En los pacientes con litiasis cálcica o de composición desconocida, se debe determinar en orina de 24 horas la creatinina, calcio, fósforo, oxalato, ácido úrico, citrato, magnesio, sodio, potasio y diuresis total. En los pacientes con litiasis no cálcicas, los estudios de evaluación metabólica son más simples y, se deben dirigir a los factores causales específicos. En los pacientes con cálculos de ácido úrico se determina en sangre la glucosa, creatinina, HDL-colesterol, triglicéridos y ácido úrico, mientras qué en el sedimento de orina, el pH, densidad urinaria y parámetros automatizados. En orina de 24 horas, la diuresis, creatinina y ácido úrico.
En pacientes con cálculos de cistina en sangre se determina creatinina y en el sedimento de orina, el pH, densidad urinaria y cociente cistina/creatinina en orina de micción o bien, en orina de 24 horas la diuresis y la determinación cuantitativa de cistina [17].
Para el despistaje de la enfermedad genética, en caso de sospecha, se realizarán las pruebas genéticas moleculares. No todas las enfermedades monogénicas requieren estudio genético, como en el caso de la cistinuria en que la determinación de la aminoaciduria en orina es diagnóstica y, en la que conocer el genotipo no cambia nuestra actitud terapéutica ni tampoco el pronóstico de la enfermedad [18] [19] [20].
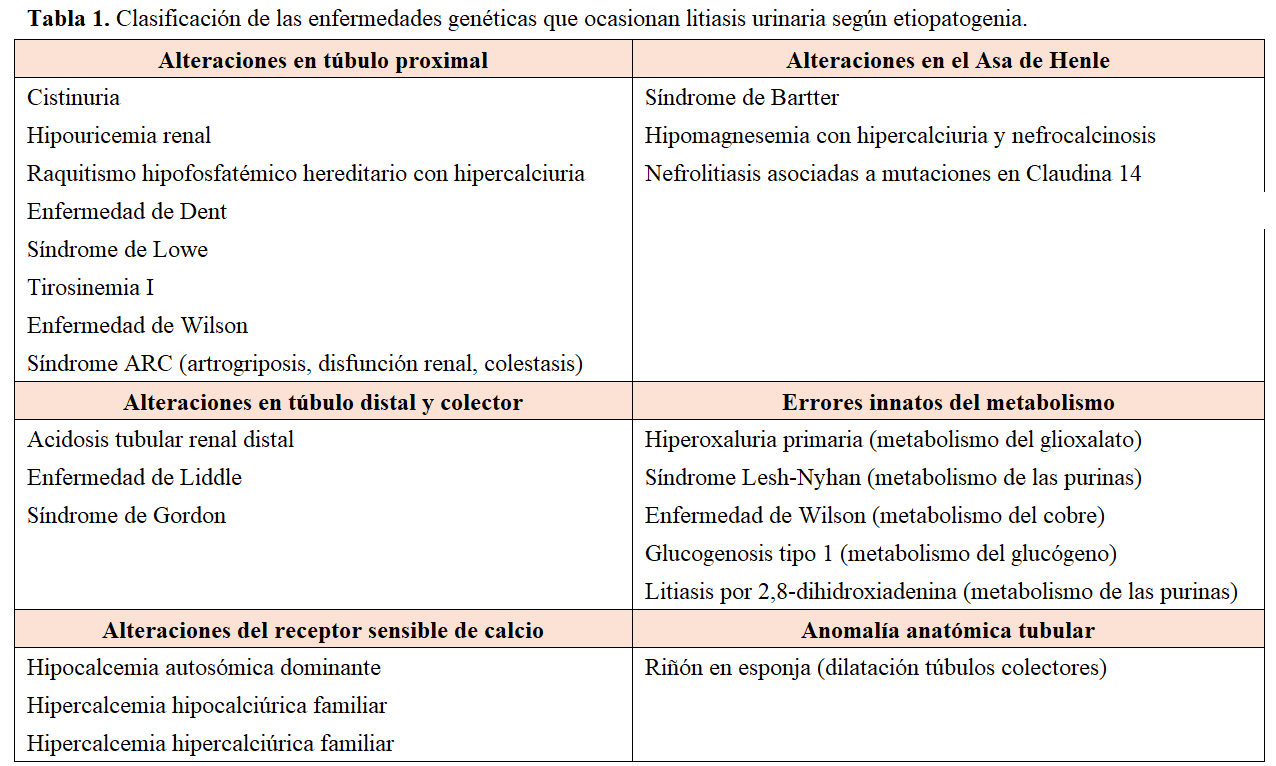
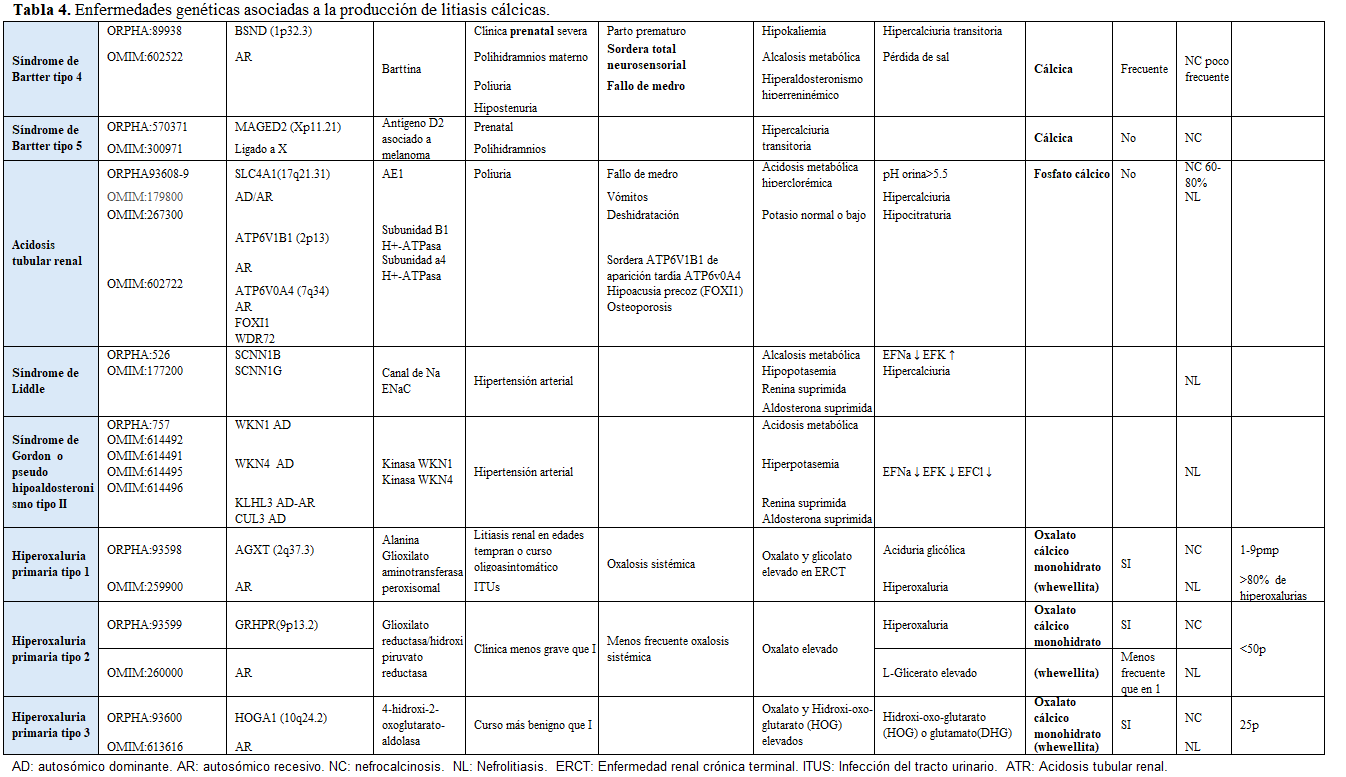
Clasificación de las principales enfermedades genéticas asociadas a la litiasis urinaria.La clasificación de estas enfermedades se puede realizar de distintas formas. Una de ellas, de elaboración propia, atiende a sí el defecto se debe a una alteración en la función del túbulo renal, a un error innato del metabolismo o a una alteración anatómica. Dentro del grupo de las alteraciones tubulares, se subclasifican según el segmento del túbulo renal afectado (Tabla 1). Esta clasificación simplifica y resume de forma práctica estas patologías [21].
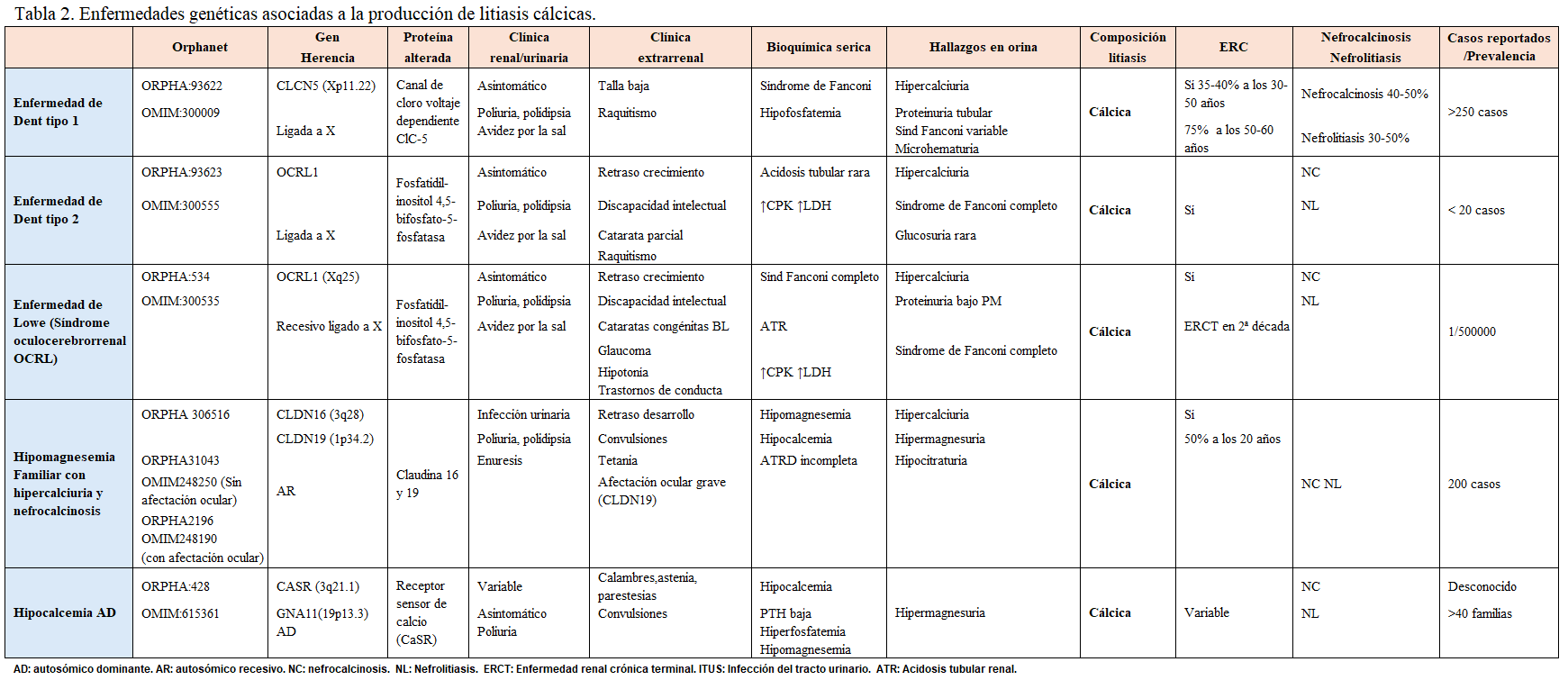
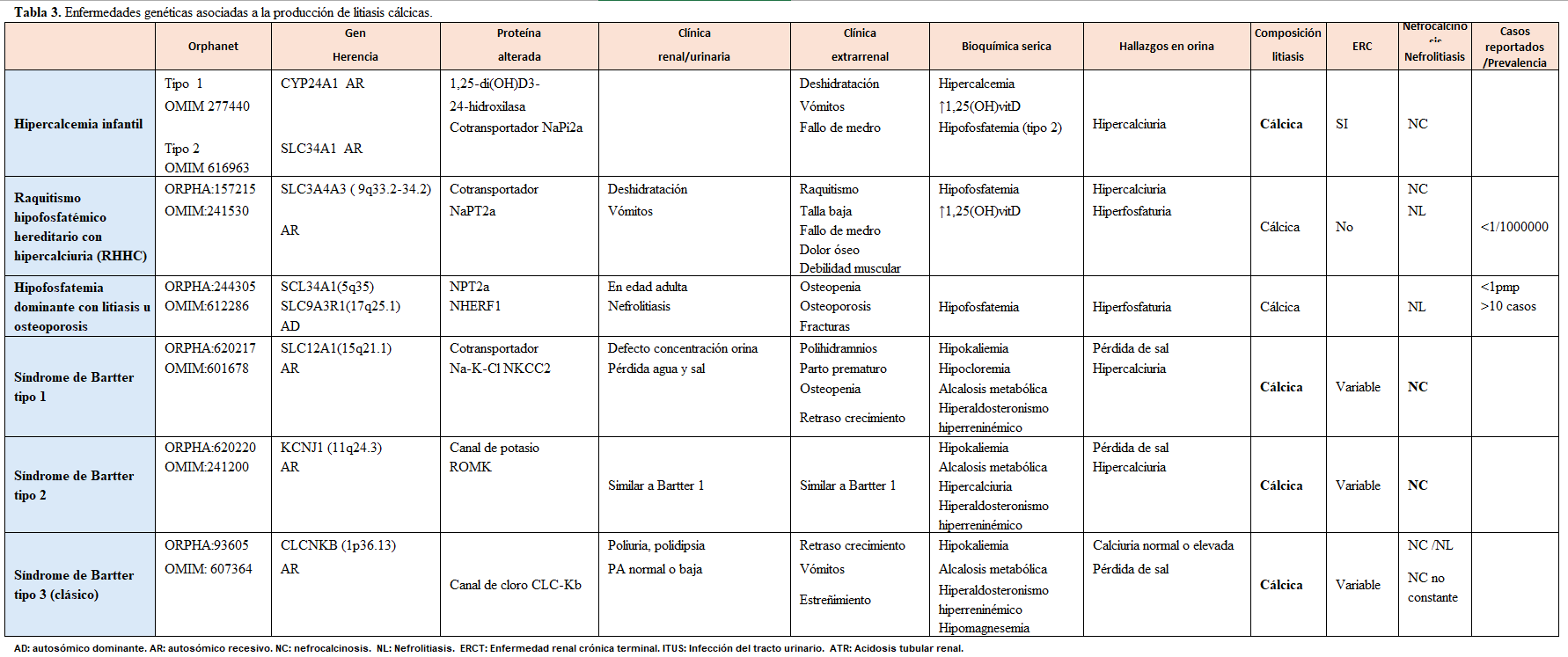
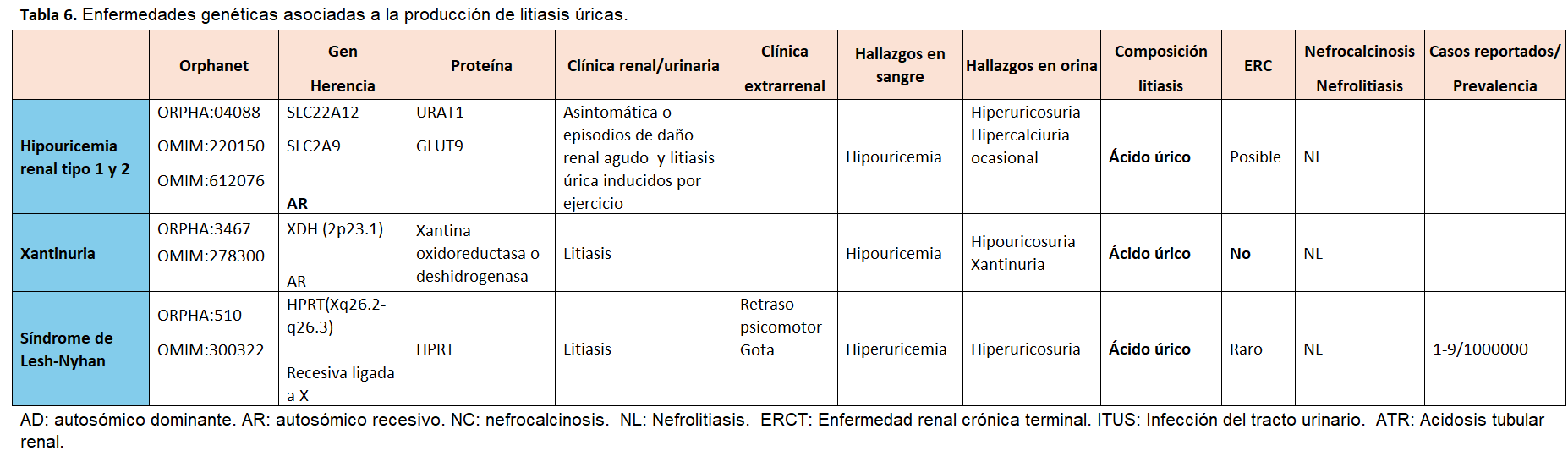
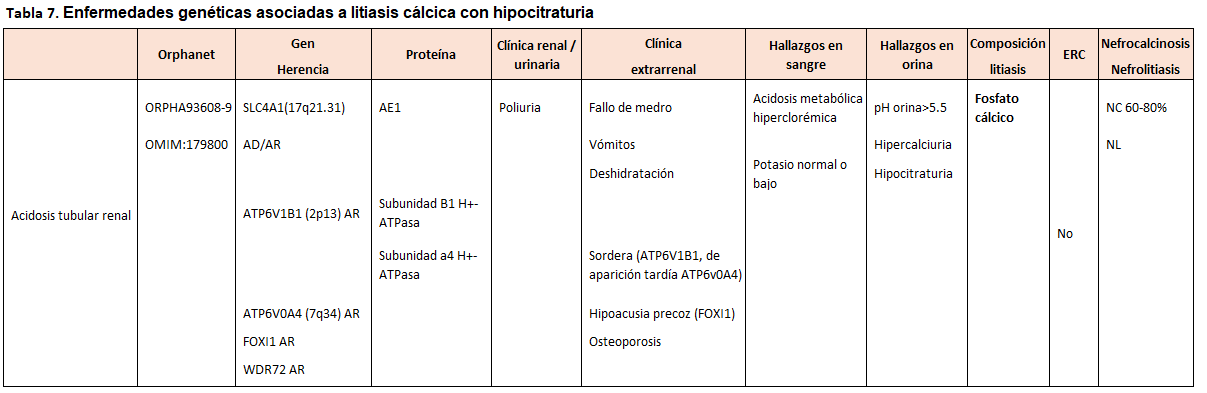
Otra forma de clasificarlas es según el tipo de litiasis que producen, tal y como se muestra en la (Tabla 2) (Tabla 3) (Tabla 4) (Tabla 5) (Tabla 6) (Tabla 7) (Tabla 8) (Tablas 2, 3, 4, 5 y 6). Se describen las principales enfermedades genéticas de acuerdo con el tipo de litiasis (cálcica, úrica o de cistina). En esta misma tabla, se detallan las características genéticas, clínicas y de laboratorio de cada una de ellas [22] [23] [24] [25].
La hipercalciuria y la hipocitraturia son las alteraciones metabólicas más frecuentes en el paciente litiásico. La hipercalciuria idiopática es la causa más frecuente, es una entidad multifactorial en la que hay una interacción compleja entre ambiente y factores genéticos individuales que regulan los procesos de absorción intestinal, excreción renal y resorción ósea que conllevan a la excreción elevada de calcio en orina. Bajo el término de hipercalciuria idiopática se han ido identificando entidades de causa monogénica. Una mejor caracterización se podrá obtener a medida que se vayan realizando mayor número de estudios genéticos [26].
Factores que orientan a patología monogénica en un paciente con litiasis urinariaEn pacientes con historia litiásica, pueden existir una serie de factores que orienten hacia una posible alteración monogénica. Es importante pensar en ellos, dado que se puede instaurar un tratamiento preventivo precoz y así evitar nuevos eventos litiásicos, prevenir o retrasar la posible insuficiencia renal o desarrollo de nefrocalcinosis y, actuar sobre las complicaciones extrarrenales [27]. Estos factores se recogen en la (Tabla 9).
Edad de inicioLa litiasis infantil es poco frecuente, constituye el 1% de los cálculos urinarios y, de éstos, un 40% tiene antecedentes familiares sugiriendo una alteración genética. Por ello, ante un primer episodio litiásico en la época infantil, se debe pensar en una posible causa genética, sobre todo cuanto menor es la edad del paciente. Sin embargo, dada la variabilidad clínica de muchas de estas entidades como la cistinuria o la hiperoxaluria primaria tipo 1, pueden diagnosticarse en edades más tardías. En la hiperoxaluria primaria tipo 1, se puede observar una nefrocalcinosis bilateral e insuficiencia renal con debut en lactantes o aparecer en la edad adulta con un cálculo simple y función renal normal [28]. En el caso de la cistinuria, la edad media para el primer cálculo renal tiene lugar antes de los 15 años, pero hay casos de debut tardío, en la edad adulta.
Historia familiar y consanguinidad de los padres.La influencia de la carga genética parece jugar un importante papel en la litiasis. Hasta un 45-55% de los casos presentan antecedentes familiares. La consanguinidad tiene también un papel relevante, así en zonas en las que el índice endogámico es mayor (como las Islas Canarias), la hiperoxaluria primaria es responsable del 10% de casos de enfermedad renal crónica en niños, muy por encima del resto de Europa (<0,5%) [29]. Cuando se diagnostica un defecto genético en un paciente debemos hacer el screening para el mismo trastorno al resto de miembros de la familia, sobre todo a los hermanos. Esto es especialmente importante cuando estudiamos a algún familiar como posible candidato a donante vivo [30]. De esta manera se pueden diagnosticar de forma precoz antes de que aparezcan los síntomas de la enfermedad e instaurar un tratamiento adecuado. Sin embargo, en la enfermedad monogénica, la ausencia de la enfermedad en otros familiares no descarta que la litiasis urinaria se deba a ella.
Enfermedad Renal Crónica (ERC)La enfermedad renal terminal (ERT) es rara en pacientes litiásicos crónicos. Normalmente está relacionada con factores metabólicos como la obesidad e hipertensión arterial, con historia de pielonefritis, episodios repetidos de obstrucción del tracto urinario y procedimientos quirúrgicos urológicos [31] [32]. Otra causa de ERC es la monogénica. Esta se debe sospechar ante pacientes litiásicos con ERC independientemente de la edad. En entidades como la hiperoxaluria primaria tipo 1, la hipomagnesemia familiar con hipercalciuria o la enfermedad de Dent lo habitual es que la ERC terminal aparezca en edades tempranas, pero dada la heterogeneidad genética, bioquímica y fenotípica de estas entidades, hay casos de inicio tardío en edad adulta avanzada [33]. La insuficiencia renal no siempre va precedida de síntomas o episodios de litiasis urinaria en pacientes con enfermedades renales hereditarias y, en ocasiones, estos pacientes no presentan ninguna clínica con una progresión lenta a ERT o incluso, en algunos casos, el diagnóstico se hace ante una recurrencia de la enfermedad litiásica en el trasplante renal. En el caso de la cistinuria la evolución a ERT es rara [34].
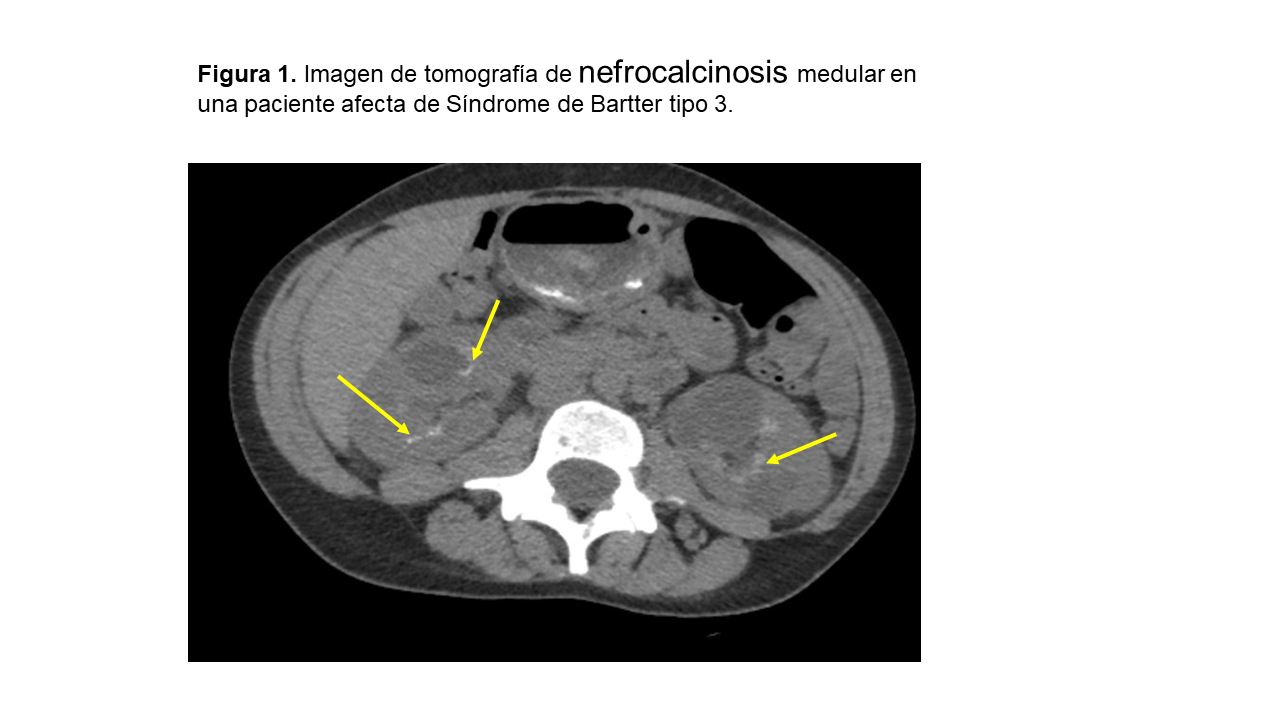
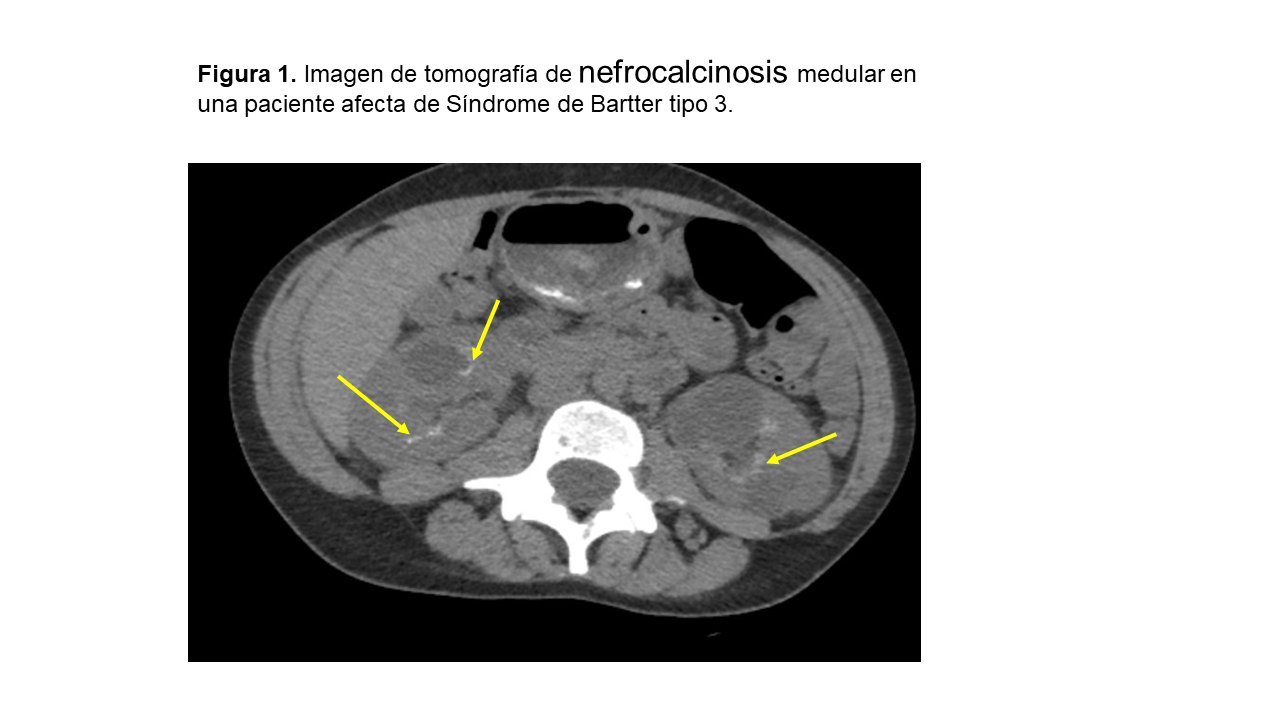
NefrocalcinosisLa nefrocalcinosis se trata de un depósito de oxalato y fosfato cálcico en el tejido renal (Figura 1). La causa más frecuente en la edad adulta es el hiperparatiroidismo primario, seguido de la acidosis tubular renal distal (dATR) (tubulopatía que más frecuentemente se asocia a nefrocalcinosis) y el riñón en esponja medular. En niños, las causas más importantes de nefrocalcinosis son las tubulopatías y los errores innatos del metabolismo que se manifiestan en edades muy tempranas [35]. La nefrocalcinosis se clasifica en medular (97%) o cortical (3%).
La hipercalciuria se considera el factor de riesgo más importante, pero hay que descartar otros desórdenes metabólicos como la hipocitraturia o la hiperoxaluria.
La nefrocalcinosis suele manifestarse de forma asintomática con una evolución lenta y progresiva. La evolución a ERT es muy variable.
Las tubulopatías que se asocian en mayor medida a nefrocalcinosis, además de la dATR, son la hipomagnesemia familiar con hipercalciuria y nefrocalcinosis, la enfermedad de Dent y la hiperoxaluria primaria [36].
En la acidosis tubular renal distal tipo 1(dATR) los pacientes afectados tienen una acidosis metabólica hiperclorémica con anión-gap normal y orina alcalina (pH >5.3). La prevalencia de nefrocalcinosis alcanza el 60-80% de los casos. La enfermedad de Dent afecta fundamentalmente a varones y debemos sospecharla ante nefrocalcinosis de inicio precoz asociada con hipercalciuria y proteinuria de bajo peso molecular. La hipomagnesemia familiar con hipercalciuria y la nefrocalcinosis evoluciona de forma casi invariable hacia la enfermedad renal crónica.
El riñón en esponja medular es una malformación congénita de los conductos colectores terminales que suele cursar de forma asintomática. Se asocia a nefrolitiasis y nefrocalcinosis, pero la evolución a ERT es poco frecuente.
Carga litiásica y RecurrenciaHasta el 50% de los pacientes que presentan un episodio de litiasis, volverán a desarrollarlo a los 5 años sin un tratamiento preventivo adecuado. Las entidades genéticas se relacionan con una mayor actividad litiásica como es el caso de la cistinuria, en la que la aparición de cálculos suele ser antes de los 15 años, la litiasis es bilateral en más del 75% de los casos y, la tasa de recurrencia es superior al 60%. Esta patología exige un cumplimiento terapéutico muy estricto y suele asociar poca adherencia por parte del paciente. En general, la presencia de litiasis múltiples bilaterales es sugestiva de enfermedad de causa hereditaria.
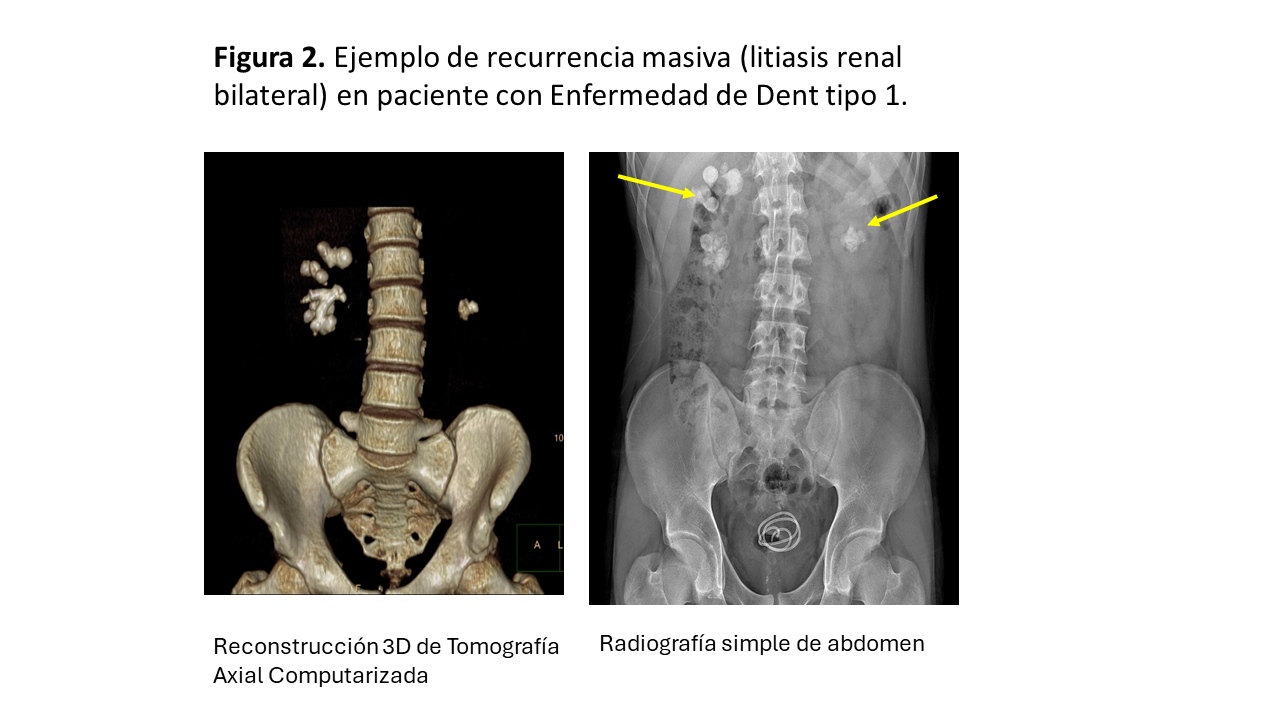
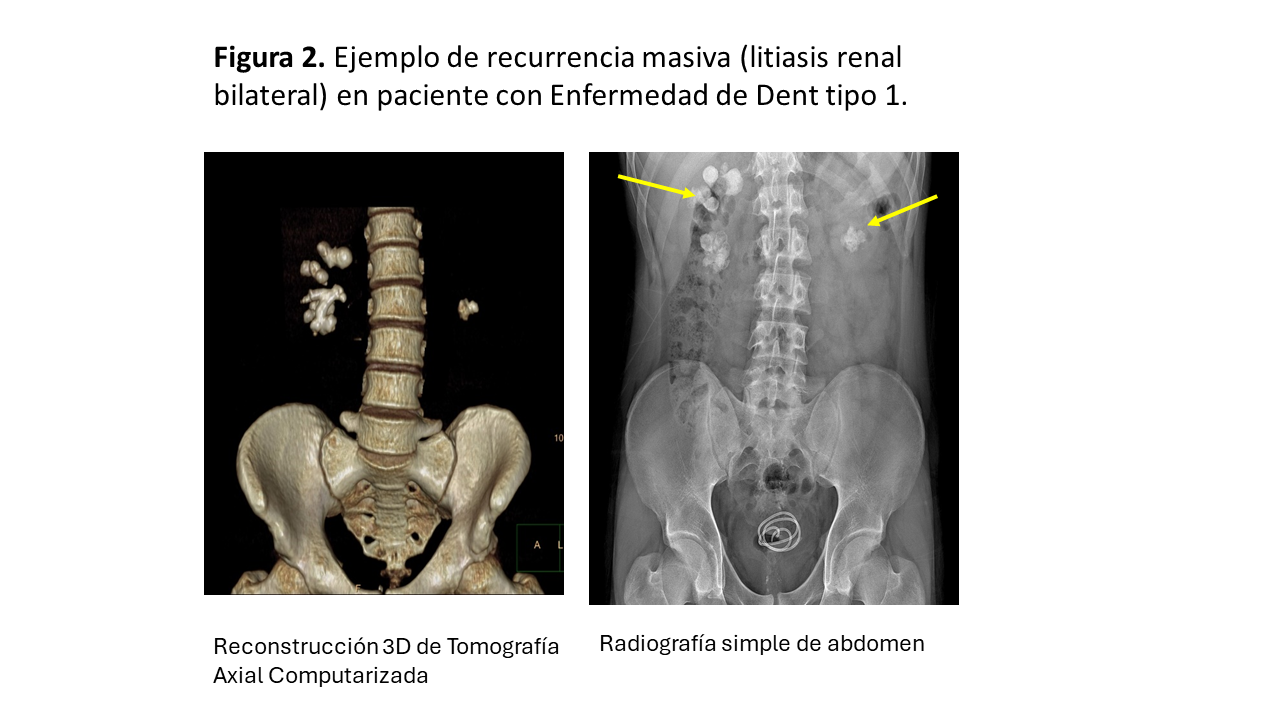
La (Figura 2) muestra un ejemplo de recurrencia masiva (litiasis renal bilateral) en paciente con Enfermedad de Dent tipo 1.
Alteraciones tubulares/extrarrenalesCiertas alteraciones analíticas sanguíneas y urinarias son orientativas de algunas alteraciones tubulares. Así, una hipomagnesemia con hipercalciuria en un paciente con nefrocalcinosis es altamente sugestivo de hipercalcuria familiar con nefrocalcinosis y, si se acompaña de una alcalosis metabólica, es sugestivo de un síndrome de Bartter. Un paciente con hipercalciuria, hipofosfatemia y proteinuria de bajo peso molecular orienta a una enfermedad de Dent. Ante un paciente con hipercalciuria, hipocitraturia, acidosis metabólica hiperclorémica y orina alcalina en un paciente con litiasis debemos descartar una acidosis tubular distal.
En ausencia de malabsorción de grasas e hiperoxaluria entérica o dietética, una elevación de oxalatos en orina es sugestivo de hiperoxaluria primaria.
La coexistencia de manifestaciones extrarrenales, sobre todo neurológicas, oculares o auditivas puede hacer sospechar en una causa genética. La sordera neurosensorial se da en algunos tipos de dATR y alteraciones oculares como la miopía, el nistagmus horizontal o las calcificaciones corneales se describen en la hipercalciuria familiar con hipomagnesemia y nefrocalcinosis. La alteración neurológica es grave en enfermedades como el síndrome de Lowe y la enfermedad de Lesh-Nyhan.
En las (Tabla 5) (Tabla 6) y (Tabla 7) se describen las alteraciones que caracterizan a cada entidad.
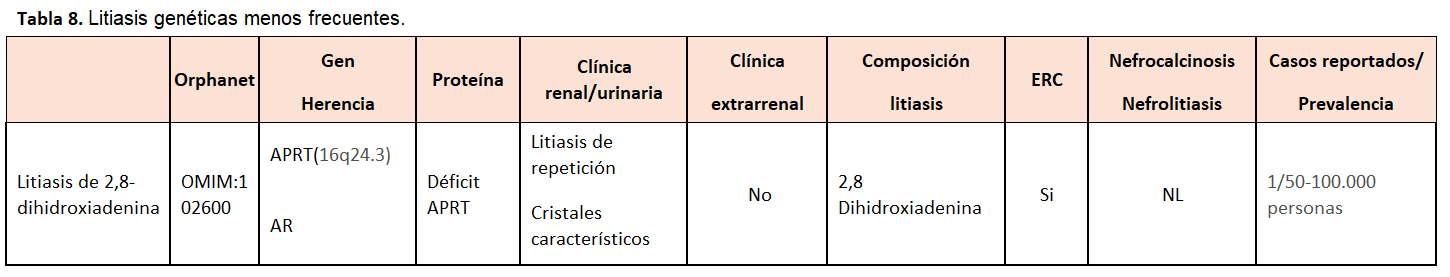
Composición de la litiasisEs el primer paso para la evaluación metabólica de un paciente formador de litiasis. Un adecuado análisis de la litiasis es fundamental para iniciar el proceso diagnóstico de la alteración metabólica subyacente. En ocasiones, la presencia de cristales en la orina nos puede orientar hacia alguna alteración metabólica. Así, los cristales hexagonales en orina son típicos de la cistinuria y, los cristales de 2,8 dihidroxiadenina, son característicamente esféricos o granulares en forma de abanico, birrefringentes a la luz polarizada y de color marrón.
Ciertas alteraciones metabólicas se asocian a tipos específicos de cálculos. Así, los cálculos renales en la hipercalciuria idiopática suelen ser de oxalato cálcico dihidrato (whedellita) o mixtos por fenómenos de enucleación heterogénea (núcleo de urato).
Las litiasis más frecuentes de fosfato cálcico son de dos tipos: carbonato de apatita (cristaliza a pH>6.8 y puede estar asociada a infección) y brushita (cristaliza a pH 6.5-6.8) en concentraciones de calcio urinario >300mg/día y fosfato>1000mg/día. Las causas más frecuentes de litiasis de carbonato apatita son la hipercalciuria, la acidosis tubular primaria o las infecciones del tracto urinario. La litiasis de brushita se asocia a hiperparatirodidismo primario o acidosis tubular primaria [37].
Los cálculos de oxalato cálcico monohidrato (whewellita) (Figura 3) son debidos a hiperoxaluria primaria o secundaria (entérica o dietética).
En la enfermedad de Dent podemos encontrar litiasis de oxalato cálcico o de fosfato cálcico.
La litiasis de cistina es patognomónica de la cistinuria (Figura 4), al igual que los cálculos de xantina en la xantinuria.
La litiasis por 2,8-dihidroxiadenina se produce por un trastorno del catabolismo de las purinas de herencia autosómica recesiva, por el déficit de la enzima APRT, que condiciona una alteración del paso de adenina a AMP y acúmulo renal de 2,8-dihidroxiadenina. Puede manifestarse tanto por insuficiencia renal crónica progresiva, por daño intersticial crónico (nefropatía por cristales), litiasis de repetición o insuficiencia renal aguda [38, 39).
La Xantinuria es un trastorno del metabolismo de la purina causado por un déficit hereditario del enzima xantina deshidrogenasa/oxidasa y, se caracteriza por una concentración muy baja (o indetectable) de ácido úrico en sangre y orina y una concentración muy alta de xantina en orina. Aunque suele ser asintomática, puede detectarse urolitiasis, debido a la elevada excreción de xantina o miositis causada por la acumulación de xantina.
Tratamiento de la litiasis renal en enfermedades genéticasEl tratamiento específico de la litiasis es en la mayoría de los casos intervencionista. Según las características de la litiasis (tamaño, localización o dureza estimada por las unidades de Hounsfield medidas en la Tomografia Computarizada espectral sin contraste) los tratamientos aplicados comprenden desde la observación, tratamiento expulsivo, litotricia extracorpórea por ondas de choque (tratamiento menos invasivo indicado en litiasis no mayores a 2 cm y de baja dureza), a los tratamientos quirúrgicos endourológicos como la ureterorrenoscopia o la nefrolitotomía percutánea, para litiasis mayores a 2 cm o de menor tamaño refractarios a la litotricia extracorpórea.
Una vez resuelta la litiasis, el tratamiento debe ir dirigido a evitar la recurrencia de la enfermedad. Las recomendaciones generales son comunes a todo tipo de litiasis y consisten en una alta ingesta de agua que condicione una diuresis abundante, restricción de la sal de la dieta, reducción de la ingesta de proteínas animales hasta un máximo de 0,8 gr/kg de peso/día, alta ingesta de frutas y verduras y no restricción de alimentos ricos en calcio. El citrato y el magnesio se usan como inhibidores de la cristalización, ya que aumentan la solubilidad del calcio en orina. En caso de hipercalciurias que no respondan a las medidas generales, las tiazidas disminuyen la excreción urinaria de calcio aumentando la reabsorción de calcio en el túbulo proximal [40].
En las patologías monogénicas, el tratamiento debe ser asegurar una correcta ingesta hídrica y reponer los solutos que se pierden en exceso, para proveer inhibidores y disminuir la concentración de promotores de la cristalización urinaria. El tratamiento ha de ser precoz para evitar la progresión a insuficiencia renal. Este tratamiento debe completarse con el tratamiento específico de cada patología.
ConclusionesLa litiasis renal en ocasiones es la manifestación clínica inicial de otras enfermedades más graves, como el caso de las enfermedades genéticas comentadas.
Debe descartarse causa monogénica en un paciente adulto ante litiasis recurrente, con antecedentes familiares, que presente insuficiencia renal o nefrocalcinosis. En niños, la sospecha es ante el primer episodio litiásico. La primera medida a realizar es el análisis del cálculo, estudio metabólico en sangre y orina, analizando los promotores e inhibidores de la formación de la litiasis, seguido, si es necesario, del estudio genético específico según la sospecha clínica.






{kind=link}
{kind=link}
{kind=link}
{kind=link}